客户文章丨GUT MICROBES嗜粘蛋白阿克曼氏菌与非酒精性脂肪肝炎
下一代益生菌嗜粘蛋白阿克曼氏菌(A.嗜粘蛋白菌)对非酒精性脂肪性肝(NAFLD)具有治疗潜力,特别是在非酒精性脂肪性肝炎(NASH)的炎症阶段。然而,A.嗜粘蛋白菌在NASH的预防作用未知。浙江大学医学院附属第一医院传染病诊治国家重点实验室王保红团队于2023年在Gut Microbes期刊上发表“Akkermansia muciniphila inhibits nonalcoholic steatohepatitis by orchestrating TLR2-activated γδT17 cell and macrophage polarization” 的研究论文,补充A.嗜粘蛋白菌可预防高脂饮食诱导的NASH小鼠的肝脏炎症,其特征在于肝脏促炎性巨噬细胞(M1)和γδT、γδT17细胞减少。此外,A.嗜粘蛋白菌抑制NASH小鼠肠屏障破坏并相应下调肝脏Toll样受体2(TLR2)表达。原文链接:https://doi.org/10.1080/19490976.2023.2221485,我公司参与了该研究中短链脂肪酸酸的定量检测工作。

1、A.嗜粘蛋白菌抑制HFD诱导的小鼠NASH
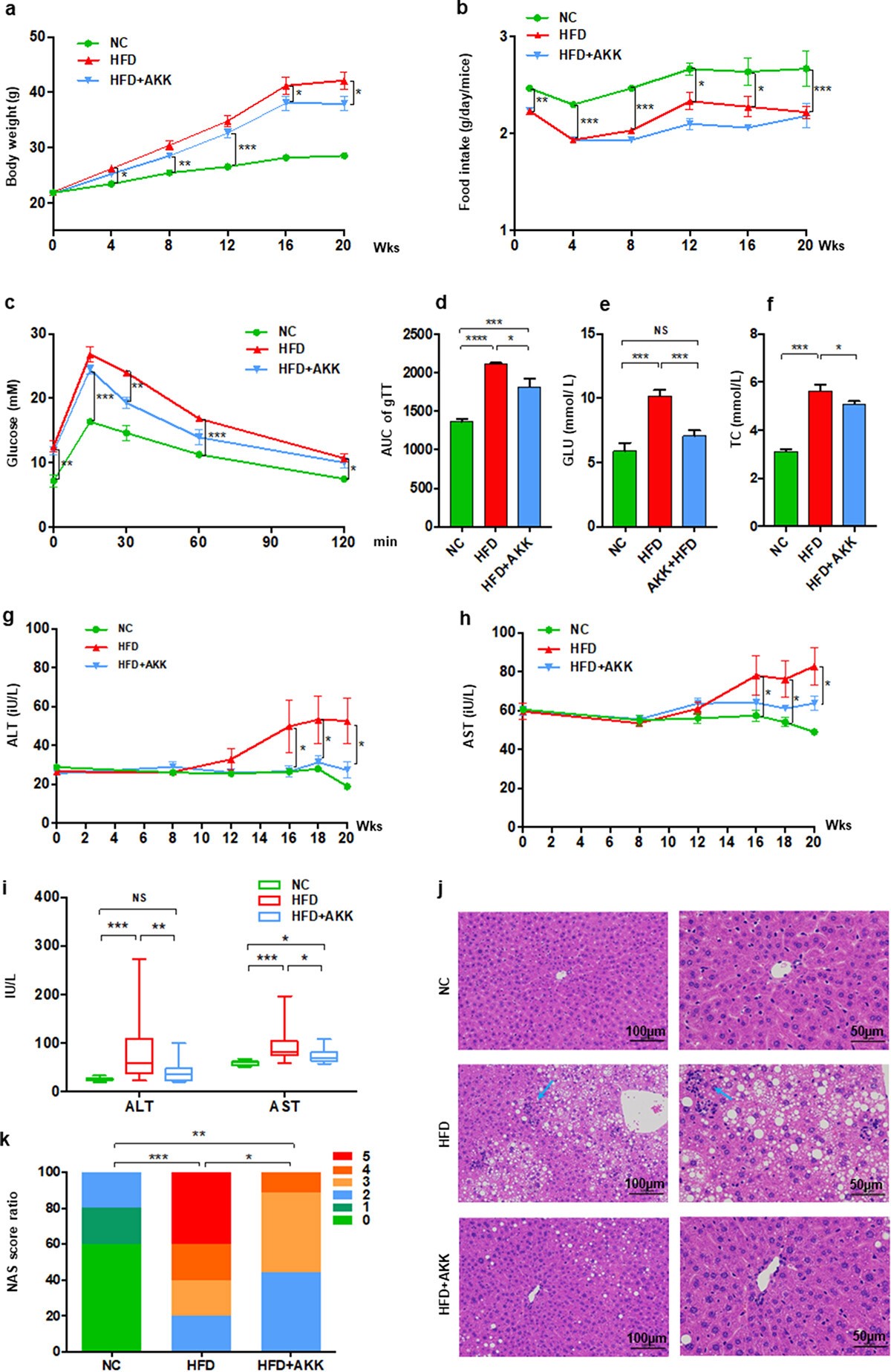
基线血清生化特征数据,包括ALT、AST、TC、TG、LDL-C、HDL-C和GLU,显示三组小鼠之间无显著差异,表明各组之间基础状况正常化。与喂食对照饲料的小鼠相比,高脂饮食(HFD)四周后小鼠体重明显增加,但摄食量减少,在实验的第16周,A.嗜粘蛋白菌明显降低HFD组小鼠的体重,但对摄食量没有影响。此外,第8周的生化检测结果表明,HFD可引起血清GLU和TC水平升高,而A.嗜粘蛋白菌没有改善。第11周,A.嗜粘蛋白菌治疗显著改善了对葡萄糖给药的反应。第12周,A.嗜粘蛋白菌改善了小鼠血清GLU的水平,但没有提高TC的水平。第18周,模型小鼠的血糖和总胆固醇均被A.嗜粘蛋白菌逆转。第20周,与对照小鼠相比,在模型小鼠中发现ALT和AST水平增加,肝脏脂肪变性和炎性细胞浸润增加,以及NAFLD活性评分(NAS)升高,在A.嗜粘蛋白菌治疗处理后明显改善。
2、A.嗜粘蛋白菌控制NASH模型小鼠γδT聚集和巨噬细胞极化
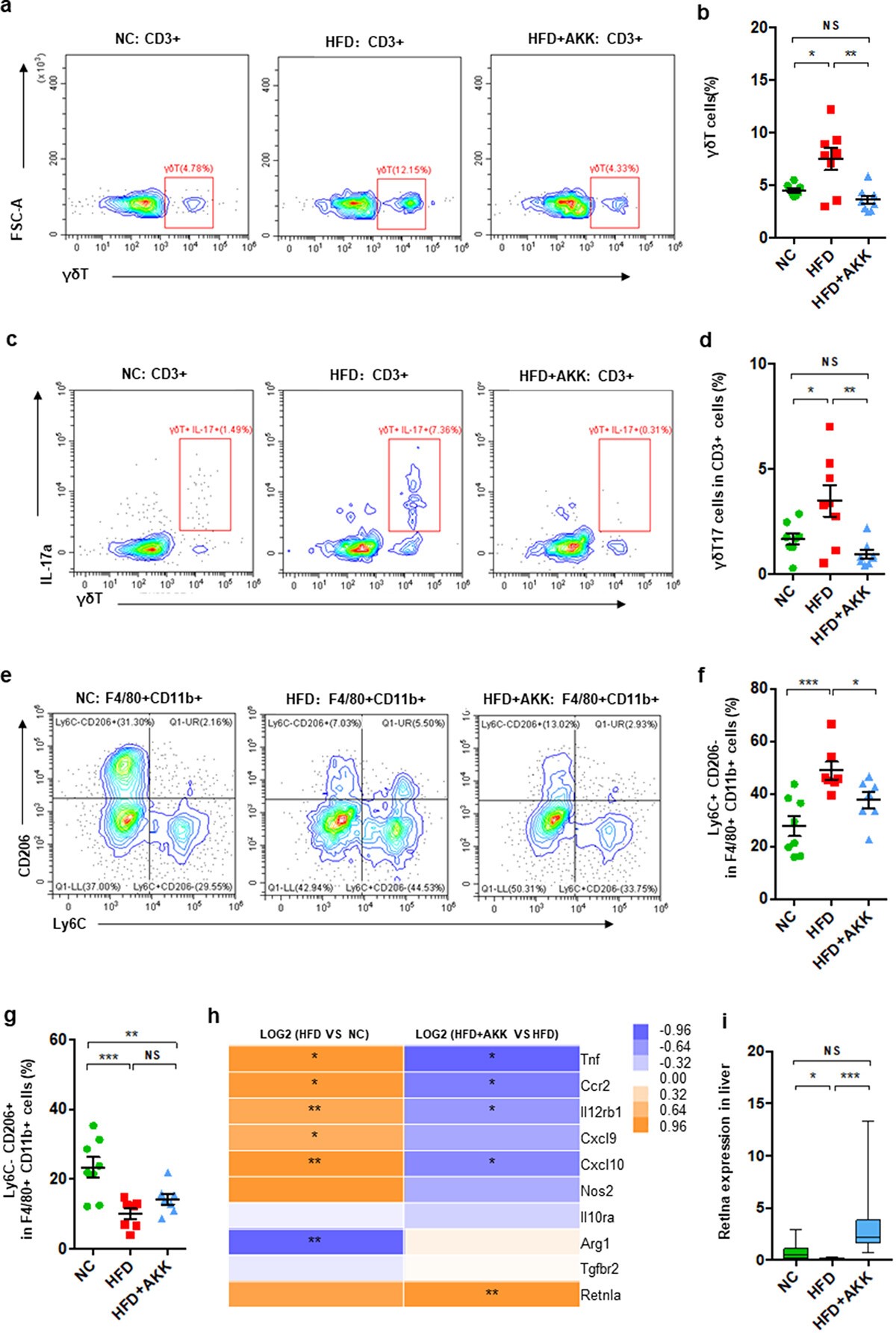
进一步研究肝脏免疫反应,在NASH小鼠中分离肝脏单核细胞并在治疗20周后进行FACS。肝脏γδT细胞,产生IL-17的γδT细胞(γδT17细胞)和促炎性M1巨噬细胞(CD11b+F4/80+Ly6C+CD206−),以及CD3+t细胞和炎性NKT细胞在HFD诱导的NASH小鼠中富集。此外,与对照组小鼠相比,抗炎M2巨噬细胞(CD11b+F4/80+Ly6C−CD206+)在NASH小鼠的肝脏中减少。重要的是,A.粘蛋白菌给药明显抑制γδT细胞、γδT17细胞和M1细胞的累积,但对其他肝脏免疫细胞无明显影响。此外,肝脏RNA-seq分析表明,HFD和HFD+AKK组小鼠的巨噬细胞极化标记发生了变化。与对照组相比,HFD小鼠肝脏中M1标记物(Tnf、Ccr 2、Cxcl 9和Cxcl 10)的基因表达增加,M2标记物(Arg 1)的基因表达减少,这部分被A.嗜粘蛋白体利用。qRT-PCR分析显示,与HFD组相比,A.嗜粘蛋白菌上调M2标志物Retnla的表达。
3、A.嗜粘蛋白菌下调NASH模型小鼠肝脏TLR2表达
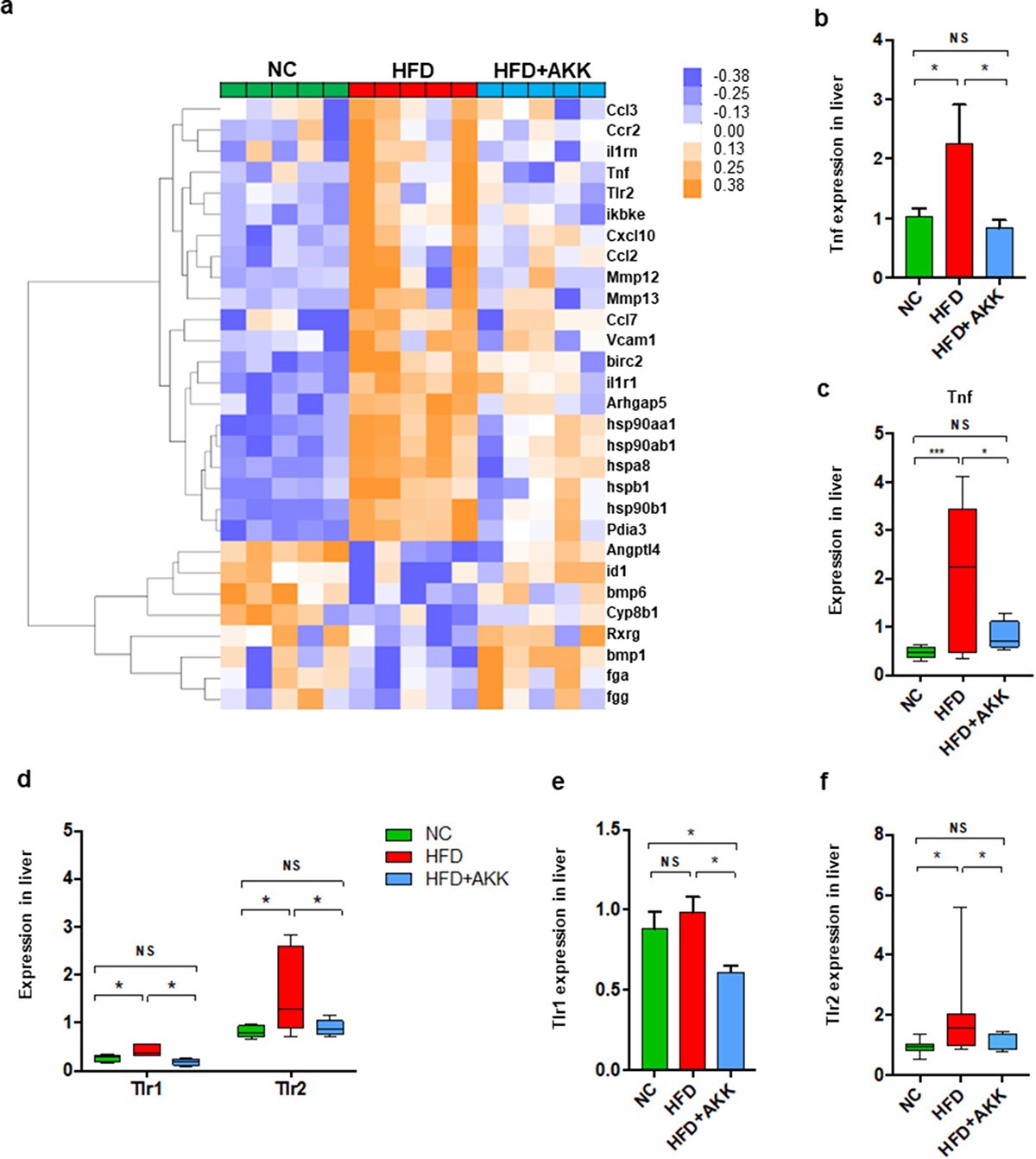
为研究细菌致病成分或免疫代谢物对肝脏免疫反应的影响,进行了RNA测序(RNA-seq)分析。HFD小鼠肝脏炎症标记物(如Tnf、Ccl2、Ccl3、Ccl7和Hsp90)的表达高于对照组,这一现象A.粘蛋白菌被部分逆转。qRT PCR分析证实A.粘蛋白菌下调HFD诱导的肝脏中Tnf表达的富集。进一步分析了识别细菌致病成分(如TLRs和NODs)或免疫代谢物(如AhR、FRAR和FXR)的受体的肝脏基因表达。HFD饲养增加了肝脏TLR1、TLR2、TLR3和TLR7的基因表达,降低了CD1d和FXR。A.粘蛋白菌仅在HFD小鼠的肝脏中逆转了TLR1/2的基因表达。因此,假设A.粘蛋白菌NASH的发生可能是由于微生物来源的TLR2信号与肝脏巨噬细胞或γδT细胞之间的相互干扰。
4、A.嗜粘蛋白菌增加NASH小鼠肠道SCFA浓度和色氨酸代谢
先前的研究表明,A.嗜粘蛋白菌产生SCFAs(例如丙酸盐和乙酸盐),参与维持肠道免疫稳态。测定了盲肠内容物中的短链脂肪酸浓度,包括甲酸、乙酸、丙酸、丁酸、2-甲基丁酸、异戊酸和戊酸。与对照组相比,HFD小鼠中甲酸、乙酸和丙酸的浓度显著降低。补充A.嗜粘蛋白菌诱导了丙酸、2-甲基丁酸、异戊酸和戊酸的富集,并导致HFD小鼠中甲酸、乙酸和丁酸增加的趋势。总SCFA浓度在HFD小鼠中降低,并通过补充逆转。相应地,HFD明显下调了SCFA受体的基因表达FFAR2(GPR43),以及A.粘蛋白菌将FFAR2表达恢复到对照组小鼠的水平。此外, HFD上调了DPPIV(使GLP-1失活的酶)的结肠基因表达,并降低了血清GLP-1水平,这些被A.嗜粘蛋白菌逆转。
除了短链脂肪酸,肠道微生物群通过代谢物(包括胆汁酸和色氨酸代谢物)的调节作用在肠道屏障保护中发挥作用。在肠道中,色氨酸可以通过三种主要途径被微生物群直接或间接代谢:吲哚途径、犬尿氨酸途径和血清素途径。基于全谱代谢组分析,发现HFD治疗减少了肠道中的大多数色氨酸代谢物,包括吲哚、褪黑激素、吲哚3-乳酸、3-羟基犬尿氨酸、吲哚3-乙酸、L-犬尿氨酸和血清素。连续给药A.粘蛋白菌部分逆转色氨酸代谢物的水平,特别是3-羟基犬尿氨酸和5-羟基犬尿氨酸。值得注意的是,A.嗜粘蛋白菌处理上调了HFD小鼠肠道中介导色氨酸降解为犬尿氨酸的酶(Ido 1、Ido 2、Tdo 2、Kynu和Kmo)的基因表达,逆转了HFD小鼠肠道中Ahr、Cyp 1b 1和IL-22的基因表达。也就是说,A.嗜粘蛋白菌还调节NASH小鼠肠道中色氨酸向犬尿氨酸途径的代谢。

5、TLR2激动剂可促进肝脏γδT细胞聚集,消除A.嗜粘蛋白菌对小鼠NASH的保护作用
进一步研究了微生物来源的TLR2与肝脏巨噬细胞或γδT细胞之间通过TLR2激动剂给药的潜在相互作用。值得注意的是,单独的脂磷壁酸(LTA)富集了饲喂正常食物饮食的小鼠中肝脏γδT细胞、TLR 2 + γδT细胞和γδT17细胞的比例,但不直接影响M1/M2细胞。LTA改变了HFD+AKK组小鼠的ALT和AST水平,使其接近HFD组小鼠的水平,但与HFD+AKK组小鼠相比,LTA对摄食量、体重、TC和TG没有影响。单独的LTA治疗药物没有改变对照组小鼠中ALT、AST、TC和TG的水平。A.嗜粘蛋白菌通过下调肠源性TLR2信号减少肝脏炎性γδT细胞。
6、γδT细胞可能通过IL-17信号与巨噬细胞相互作用
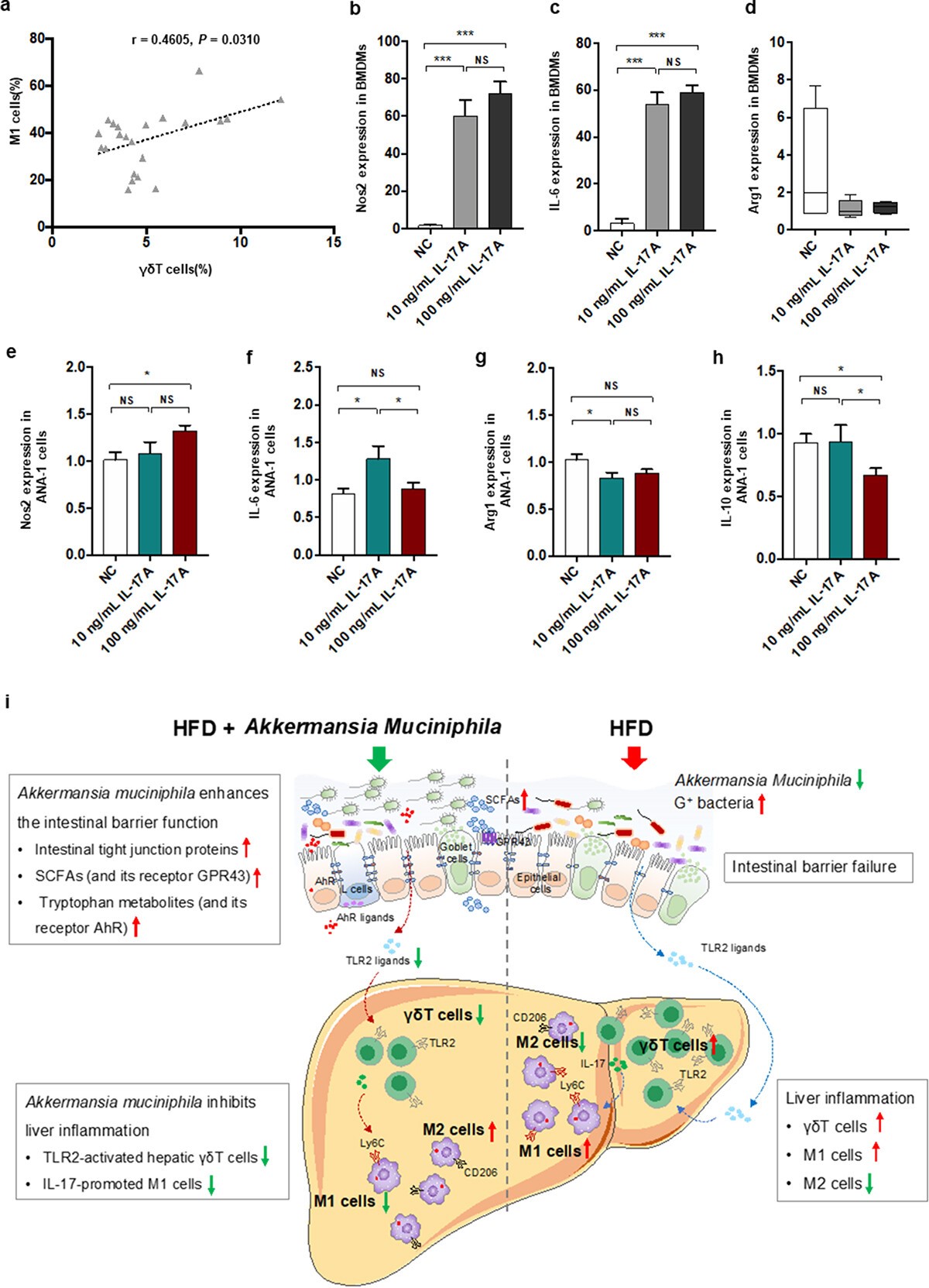
在NASH小鼠模型中,A.嗜粘蛋白菌通过调节γδT积聚和巨噬细胞极化来抑制肝脏炎症。分析测量了肝脏γδT细胞和M1细胞的水平,发现肝脏γδT细胞与NASH小鼠中的M1细胞正相关,表明γδT细胞在巨噬细胞极化中的作用。为了进一步研究γδT细胞(一种主要类型的IL−17产生细胞)是否对巨噬细胞极化有任何调节作用,在体外用重组IL− 17 A刺激巨噬细胞。用10 ng/ mL或100 ng/mL重组IL-17 A刺激骨髓源性巨噬细胞(BMDM)细胞。IL-17显著促进BMDM的M1标记物(Nos 2和IL-6)的基因表达,并轻微影响M2标记物(Arg-1)的基因表达。为了进一步验证IL−17和巨噬细胞之间的相互作用,IL−17显著促进巨噬细胞Ana−1细胞M1标志物(Nos 2和IL −6)的基因表达,并持续抑制M2标志物(Arg−1和IL−10)的基因表达。结果表明,IL-17可以促进巨噬细胞极化成促炎性M1细胞,活化的γδT细胞可以通过IL-17促进巨噬细胞极化,提示两种肝脏先天免疫细胞之间存在直接联系。
7、结论
确定了两个关键的肝脏先天免疫细胞(γδT和巨噬细胞),它们受A.嗜粘蛋白菌调节。在NASH小鼠的肝脏中,A.嗜粘蛋白菌保护肠屏障并进一步下调微生物来源的TLR 2信号传导。此外,通过给予TLR 2前药,证明了A.嗜粘蛋白菌通过下调肝TLR 2信号转导降低γδT17细胞水平。γδT细胞通过IL−17促进巨噬细胞向促炎性M1细胞极化。总体而言,A.嗜粘蛋白菌通过调节紧密连接蛋白和微生物源性代谢产物增强肠屏障,通过下调TLR 2减少肝γδT17细胞,这进一步通过IL-17调节肝巨噬细胞极化,最终抑制非酒精性脂肪性肝炎。







