文献分享丨Nat Protoc代谢组学非靶流程
代谢组学是继基因组学和蛋白质组学之后新兴发展起来的学科,是系统生物学的重要组成部分。代谢组学的概念来源于代谢组,代谢组是指某一生物或细胞在一特定生理时期内所有的小分子代谢产物,代谢组学则是对某一生物或细胞在一特定生理时期内所有小分子代谢产物同时进行定性和定量分析。色谱-质谱平台经常用于对单个生物液体或组织样本中的数百至数千种代谢物进行高灵敏且可重复的检测。
英国曼彻斯特大学Royston Goodacre团队联合人类血清代谢组学联合会(HUSERMET)对基于质谱技术的大规模血清/血浆代谢组学研究的实验流程进行了详细描述,相关研究成果于2011年7月发表于Nature Protocols,文章标题“Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry”。原文链接:https://doi.org/10.1038/nprot.2011.335。

代谢组学是系统生物学研究的核心组成,主要是对小分子量有机和无机(<1500 Da)代谢产物的整体研究。与其他“组学”平台(例如转录组学或蛋白质组学)相比,代谢物分析能够以高通量方法和较低的总成本对大样本集进行全局筛选。代谢组学分析平台包括气相色谱(GC)或液相色谱(LC/UPLC)联用质谱(MS)、毛细管电泳、核磁共振光谱、红外和拉曼光谱、电化学检测器,或直接使用MS等检测。由于哺乳动物代谢组的复杂性,没有一个单独的分析平台可以用于检测生物样本中的所有代谢物,必须使用多个分析平台来提高检测代谢物的覆盖率。GC-MS或UPLC-MS由于高通量、高精密度及低成本而广泛应用于代谢组学研究。
血清/血浆的大规模代谢组学研究
在临床代谢组学研究中,涉及的样本包括血液(血清、血浆)、尿液、脑脊液、淋巴液、胆汁、粪便、唾液、细胞、组织等,其中血液(血清、血浆)是研究最多的样本。血液在生物体内有着广泛的流动性,起着最重要的物质传输作用,包含数百或数千种代谢物,可以反应生物体最直接、最有效代谢情况。
人类的代谢表型受到了遗传因素和环境(饮食、年龄、生活方式、性别等因素)的影响。为准确有效地定义其代谢变化,需要大样本量的流行病学调查,可能涉及数千个样本(比如队列研究)。核磁共振方法由于重现性极高,样品不与仪器直接接触,具有非破坏性的优点,容易实现对数百到数千个样本的高通量分析。对于色谱-质谱平台来说,由于样品注入到分析器中,会污染仪器,存在响应差异和保留时间漂移,诸多因素限制了高分辨质谱的通量。因此,基于色谱-质谱平台,进行大规模代谢组学实验时,必须使用代表性质控样本(QC),保证数据的稳定及可重复性。代谢组学大规模研究中的通用工作流程包括:实验设计、样品制备、数据采集、数据预处理、多实验数据集成/数据分析。
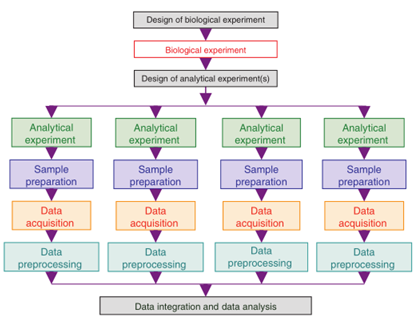
实验设计
在实际研究中,代谢物易受各种因素影响。代谢组学的研究从小规模扩展到大规模,即从单次分析样本少于100个的生物实验扩展到数月或数年内分析数千个样本。规模扩大后需要合理选择参与者(研究设计)、生物样本的收集、分析实验的设计(实验设计),以使后续的数据分析无偏向且符合目的。
在大规模的队列研究中,由于色谱-质谱系统中,代谢特征(仪器信号响应)会随着时间变化,因此,在整个分析过程中定期分析QC样品,以便为检测到的每个代谢特征提供可靠的质量保证(QA)。在后期数据处理阶段,可以通过QC校正每个样本的代谢信号,并在数据采集后和统计分析前将批量数据合并在一起。
1)样本收集与保存:血清是不加抗凝剂通过凝血和离心除去血细胞和纤维蛋白原后分离得到的上清,而血浆是从全血中通过加入抗凝剂后离心分离得到的。关于抗凝剂选择,柠檬酸盐和EDTA都可能引入干扰峰或抑制内源性分析物而影响代谢谱,因此,在收集血浆样品时首选肝素锂。制备血清和血浆后,分装储存在-80℃,避免反复冻融。
2)GC-MS应用:主要用于分析挥发性化合物或可以通过衍生化转变为挥发性的化合物。GC主要可以实现分子量范围为18-350 Da的代谢物分离,包括氨基酸、有机酸、胺、酰胺和糖等。GC-MS代谢分析常用的质谱仪为飞行时间(TOF)和四极杆质谱。样品在GC中的保留时间(常用保留指数)与经质谱EI离子源产生的特征碎片,可以帮助比对数据库进行代谢物鉴定。血清和血浆中发现的许多代谢物在GC-MS分析之前需要衍生,常用的衍生化方法是三甲基硅烷化(TMS)。
3)UPLC-MS应用:UPLC是超高效液相色谱,具有高分离度和高灵敏度等,常与TOF或傅立叶变换/轨道质谱仪联用。UPLC-MS的线性动态范围超过三到五个数量级,可以检测到浓度低于最高浓度0.01%的代谢物。常用的色谱分离法为反相色谱法,溶剂通常为甲醇或乙腈。质谱常用的离子源为电喷雾离子源(ESI),为提高代谢物覆盖率,一般进样时采用两种模式,正离子模式(ESI+)和负离子模式(ESI-)。UPLC-MS可以检测的代谢物较为广泛,包括大分子量范围(50~1500Da)代谢物,如高分子量脂质(如磷脂和甘油三酯)和非极性氨基酸。与GC-MS相比,UPLC-MS的样品制备较简单,不需要衍生,常用蛋白沉淀法。
4)血清/血浆样本的选择:血清和血浆中的代谢物种类差异不大,但并不完全相同。血浆和血清从血液中去除细胞的方法将导致产生的液体成分略有不同。血浆或血清的选择一般基于环境、设施及个人偏好。鉴于血浆和血清之间的样本成分可能存在差异,应注意确保在任何特定研究中仅使用一种样本类型。
血清/血浆样本前处理
血清或血浆样本中包括小分子代谢物和其他高分子量物质(包括蛋白质和RNA),基质较复杂。基于GC/UPLC-MS研究的血清和血浆样品,需要经过前处理后才可进样分析。常用的方法为蛋白沉淀,即添加有机溶剂或溶剂混合物以沉淀蛋白质等高分子量物质,然后通过离心分离沉淀物和含有代谢物的上清液。常用的沉淀剂为甲醇,比例为3:1(体积/体积)时可高效去除蛋白质,且步骤简单。此外,因血清和血浆含有高水平的酶,为抑制酶活性,减少对代谢物的影响,蛋白质沉淀时应使用冰溶剂或步骤在冰上进行。与UPLC-MS相比,用于GC-MS进样分析的前处理需要衍生化,步骤较多,且进样体积小(通常为1µl),容易产生误差。通常在所有样品中加入一个或多个内标物,用于校正样品处理和进样时的误差。
样本分析(数据采集)
如上所述,基于GC/UPLC-MS的样本量较小的实验,可获得重复且稳定的数据,通过质量保证程序处理多个小规模的分析实验数据,整合成为大规模代谢组实验。作者通过对GC-MS和UPLC-MS方法优化和验证后,单个分析实验的样本量宜为120个样本。此外,在GC-MS和UPLC-M的每个分析批次结束时需进行仪器维护,包括质谱仪离子源和色谱柱清洗等。
在大规模血清样本分析时,QC样本的选择及稳定可保证整个实验的准确。QC样本需与样本种类保持一致,其代谢和基质组成与研究中的生物样品相似。一般有两种QC样品,一种是混合QC,是将待研究的每个生物样品的一部分混合后制备,一种是商业QC样本。混合QC最接近研究样本,适合样本量较小的实验研究,但是不适用大规模研究。作者在血清大规模分析时,发现商用血清QC与研究样本之间存在代谢物及其相对浓度不同的情况,会丢失部分代谢信息。因此,作者建议使用来自所有受试者群体或受试者群体子集的混合QC样本,以确保最小程度的代谢信息丢失。
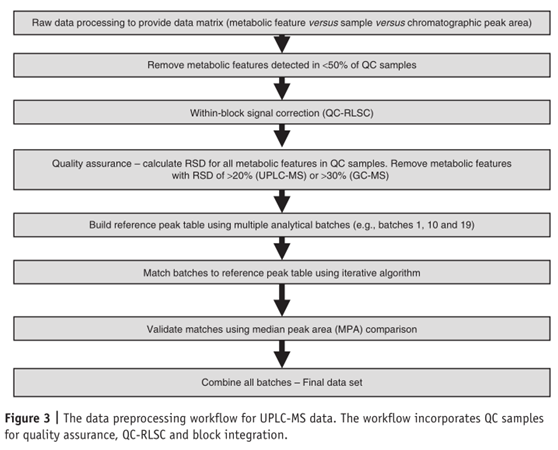
使用QC样品的原因,首先是在分析样本前可用QC平衡仪器响应、分析方法,以确保获得可再现的数据;其次是用QC提供分析数据来计算每个分析区块内的技术精度,即QA程序。对于UPLC-MS,分析时三分之二的质控样品中检测到的代谢物响应在质控平均值的20%以内,而对GC-MS,30%也可接受;第三个原因是使用QC样本来校正分析批次内及批次间的信号校正。可采用本地散点平滑估计(Locally Estimated Scatterplot Smoothing,LOESS)方法用QC进行信号校正(QC-RLSC)。即通过QC样本中相同代谢特征的信号变化,来校正周围研究样本中每个代谢特征的信号漂移。为使信号校正准确,要求在每3-5个样本中插入QC样本。
数据处理
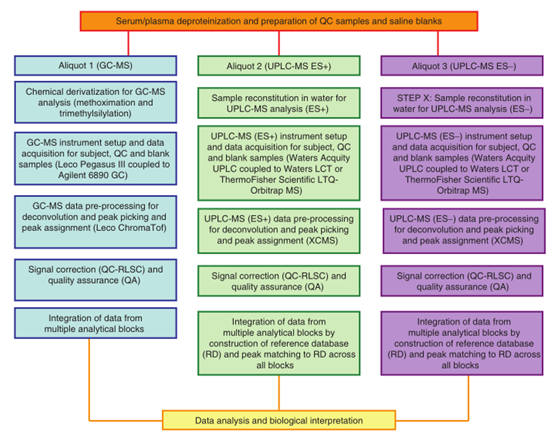
使用GC-MS和UPLC-MS采集的原始数据通常需经过预处理,以适当的格式提供结构化数据用于数据分析。使用一系列软件处理这些数据,以构建色谱峰(具有相关保留时间或指数、EI碎片质谱和/或准确质量)、峰值响应与对应样品的矩阵。这数据转化可在仪器公司自建软件中进行,如Waters MarkerLynx、ThermoFisher SIEVE、Agilent MasshHunter、Applied Biosystems MarkerView、Shimadzu Profiler AM+和LECO ChromaTOF,或在一些开源和免费软件(如XCMS89、MZmine90、Metalign91和MathDAMP92)中进行转化。
质量控制
在使用GC-MS和UPLC-MS分析时,信号强度随时间漂移的问题是长期代谢组学研究中的一个挑战。在进行数据分析时,色谱反卷积处理后,需要使用QC-RLSC将每个检测到的代谢特征归一化。使用QC样本进行QA、信号校正和数据集成的数据预处理工作流程如下图所示。详细的方法可以参考原文。
代谢物鉴定
代谢组学非靶分析中,代谢产物鉴定是最大的难题。目前,在色谱质谱采集数据的代谢组学数据集中,由于鉴定的数据库还不完善,还存在很多未识别的色谱峰。人体代谢组由内源代谢产物、外源代谢产物(例如药物、食物成分、植物化学物质)、内源和外源代谢产物的代谢产物以及来自肠道菌群的代谢产物组成,而并非所有已知的代谢物都可以购买来构建质谱库以帮助识别。一般代谢物的鉴定包括以下方法:
1)推定鉴定,将代谢特征的单个参数(如准确的质量或质谱碎片)与数据库中的代谢物进行匹配,置信度较低。
2)最终鉴定,在相同分析条件下,比对样品中存在的代谢物与标准品的两个或两个以上参数,进行匹配。
3)使用质谱库鉴定GC-MS检测到的代谢物,包括商业数据库(如NIST/EPA /NIH)、免费数据库(如Golm Metabolite Database)或实验室自建库。样本衍生代谢物的裂解质谱与文库中的裂解质谱相匹配,并以匹配概率评分。
4)基于UPLC-MS方法,代谢物的鉴定主要是根据实际得到的谱图和数据库中(如HMDB(http://www.HMDB.ca/)、KEGG(http://www.genome.jp/KEGG/)、ChemSpider(http://www.ChemSpider.com/)的谱图进行比对,根据其匹配程度获得鉴定结果。然而同一个代谢物在不同仪器平台上获得的图谱是不同的;同一个仪器平台上,不同分析条件下获得的图谱也不同等,这些都限制了代谢物的准确鉴定。
结论
文章描述了血清/血浆样品大规模代谢组学非靶分析过程中,包括样品制备、数据采集、数据预处理、信号校正和数据集整合过程等,如下图所示。这为代谢组学非靶大规模研究提供了标准的操作规程,有助于人类的代谢组学研究发展。