经典重读丨Anal Chem代谢组学分析流程
自1999年代谢组学诞生以来,每年发表的代谢组学研究的文章数量都在不断增加,在疾病诊断、药物研发、食品安全、环境科学等诸多领域都有极其广泛的应用,我司精心挑选了一批代表性的科学文献,逐一重读,带你用一杯茶的时间,理解近20年来的代谢组学技术进展和重要应用。

在“组学”技术中,代谢组学的分析发展产生了深远影响,由于代谢物的不同理化性质及浓度范围,需要更加强有力的分析策略。来自西班牙圣帕布洛大学(Universidad San Pablo CEU),波兰格但斯克医科大学(Medical University of Gdańsk)和波兰比亚韦斯托克医科大学(Medical University Of Białystok)的研究者们于2020年在Analytical Chemistry上发表代谢组学分析流程最新进展综述,具体内容包括(1)样品收集和制备(2)分析工具以及(3)数据挖掘策略和化合物鉴定的新趋势。文章标题“Recent Developments along the Analytical Process for Metabolomics Workflows”,原文链接:https://doi.org/10.1021/acs.analchem.9b04553。
一、样品收集
制定和优化样本收集和处理的标准操作程序对于获得可靠数据以及在涉及不同生物样本库分析的流行病学规模的代谢组学研究中至关重要。
1.血液、血浆和血清
血液、血浆和血清是代谢组学分析使用最广泛的基质,用于疾病研究以揭示潜在的生物标志物。尽管目前已有标准化的血液样本采集方案,但针对非靶向代谢组学研究的最佳策略仍在争论中。一些研究对血浆和血清样本进行了基于LC-MS的靶向代谢组学分析,探究样本类型和收集管选择对代谢物绝对浓度的影响,发现血清样本中的代谢物浓度高于柠檬酸盐血浆和EDTA血浆。此外,如果储存不当,许多代谢物水平特别是一些脂类,如溶血磷胆碱和鞘磷脂,在样本采集后30分钟内会发生变化。
近年来发展了一些新的采样技术,如体积吸收微量采样技术(Volumetric absorptive microsampling,VAMS),用于收集固定体积(10 μL,20 μL和30 μL)包括血液在内的各种生物体液,其对靶向和非靶向代谢组学研究的适用性仍在评估中。为了克服传统静脉穿刺术的主要缺点,开发了使用友好且无痛的触摸激活静脉切开术(Touch-activated phlebotomy,TAP),对45种代谢物的靶向LC−MS分析表明,45种代谢物中有39种,包括大多数氨基酸、胆汁酸、N,N-二甲基-L-甘氨酸、三甲胺N-氧化物、葡萄糖、乳酸和酰基肉碱,在来自传统血液采集和TAP采集的样品中具有可比性,而半胱氨酸、还原型谷胱甘肽、琥珀酸和尿酸的水平有显著差异。在最近的一项研究中,采用了干血斑甲醇提取方法用于高通量、基于芯片的nano-ESI-MS分析,耗时短,溶剂消耗最少,无过载情况。
在常规临床环境中正确收集的任何血液样本,包括最初为其他目的收集的血液样本,都可能被用于差异分析的代谢组学。然而,重现性在样品收集和制备中至关重要。因此,强烈建议遵循可靠的标准操作规程。
2.尿液
就稳定性而言,尿液样本应始终保存在低于-20°C的温度下。考虑临床条件,尿样可以在−20°C或更低的温度下保存少于30天,长期保存建议−80°C。
3.组织
使用组织样本的代谢组学的第一个挑战是样本的异质性,对于特定区域的代谢,必须特别小心。大脑是最复杂的器官之一,由于许多病理现象仅限于特定的区域。例如,神经原纤维缠结是阿尔茨海默病的主要特征之一,主要位于海马体和大脑的皮质/边缘区域。其他异质性组织如肝和肺。
第二个挑战是通过使内源性酶失活来有效地抑制正在进行的代谢反应。尽管有不同的样本保存方法,但最普遍的方法是在液氮(-196℃)中速冻。组织样本采集的一个内在因素是许多代谢物的变化,因为合成代谢反应在死后到组织收集的时间段内停止,分解代谢反应不会。代谢组学分析不仅有助于揭示个体死亡后组织内可能的变化,还有助于法医学、癌症和移植研究。
4.粪便
在了解肠道微生物对健康的广泛影响的同时,粪便代谢组学也引起了越来越多的关注,因为粪便是一种非侵入性的基质,代表了肠道中消化过程的最终产物。对于人体研究,志愿者可以使用消毒的广口塑料袋或容器(例如Fecotainer)等工具自行收集粪便样本,且应在采集样本前接受指导,以避免粪便被水、尿液或卫生纸污染。在任何生物基质的代谢组学研究中,样本储存都是至关重要的,粪便也不例外。因此,在采集样品后,必须立即将样品放置在密封、绝缘的容器中,并在2小时内送到实验室,在−80℃下储存,防止微生物发酵。
多个因素包括饮食暴露、运动、药物、一天中的时间和大便本身的异质性,将影响基于粪便的代谢组学研究结果。因此,最佳策略需要将新鲜样本直接在运输容器中匀质,然后在将样本放置在−80°C之前等分为多份,避免冻融循环。由于粪便粉末具有吸湿性,称量时必须小心。
5.细胞
细胞单细胞培养是生物材料的重要来源,是生物体液和组织的替代品。代谢组学领域对代谢调节的研究一直集中在这类样本上。然而,关于细胞代谢组学的最佳工作流程,如收集、淬灭和代谢物提取,缺乏共识。在代谢组学研究中,胰酶消化法和刮取法是两种广泛使用的获取贴壁生长细胞的方法。有研究在比较了刮取法和胰酶消化法收集贴壁细胞(HeLa和MCF7)以及冻融循环和玻璃微珠作为细胞裂解策略后得出结论:刮取和冻融循环相结合是利用同位素标记LC-MS分析贴壁哺乳动物细胞代谢组学的最佳方法。另一个可能对细胞代谢组学研究有重大影响的关键因素是传代次数。Abdul-Hamid等人用物理刮刀和胰酶消化的方法收集三种传代次数(9,12和14)的RAW 264.7细胞,并通过1H NMR分析。结果表明,传代次数越多,三羧酸循环和糖酵解所涉及的氨基酸和代谢物水平越低。另一方面,收集方式对甲硫氨酸和半胱氨酸途径以及甘油磷脂代谢也有影响。因此,作者提出,传代次数较少(<9P)的胰酶消化的原始细胞更适合于基于1H NMR的代谢组学。
尽管在前面描述的研究中使用了胰酶消化,但有报道发现在细胞胰酶消化过程中发生了代谢物渗漏,因此必须谨慎使用这种方法,而这种渗漏主要是由于收集过程本身的破坏性。
基于细胞的代谢组学研究的一种潜在的最佳工作流程可能包括首先通过荧光激活细胞分选(Fluorescence-activated cell sorting,FACS)分离感兴趣的细胞类型,然后在细胞裂解之前淬灭细胞代谢。然而,细胞中代谢物水平可能在细胞分选到淬灭的时间段内发生改变。LluFrio等人发现,FACS诱导星形胶质细胞氧化应激,改变了细胞的氧化还原状态,改变了近一半检测到的代谢物的信号强度。主要的发现是改变了谷胱甘肽二硫化物(GSSG)与谷胱甘肽(GSH)、NADPH与NADP+、NAD+与NADH的比率,同时发现参与TCA循环、糖酵解和磷酸戊糖途径的氨基酸、酰基肉碱等代谢物水平的降低。相反,FACS-星形胶质细胞的尿苷和肌苷水平升高。作为这项研究的继续,Binek等人进行了多平台(液相色谱-质谱、气相色谱-质谱和毛细管电泳-质谱)代谢组学分析,以研究流式细胞仪对C57BL/6小鼠腹膜巨噬细胞代谢组的影响。作者评估了免疫染色、FACS以及这两种技术结合的效果,并与对照组的细胞进行了比较。结果表明,FACS至少影响10%检测的代谢物,包括甘油磷脂、脂肪酰基、脂肪酸酯、氨基酸(及其衍生物)、甘油脂和鞘脂。这些变化与FACS在细胞代谢组中诱导的物理应激有关,包括诱导炎症样状态,涉及Ca2+信号和磷脂酶A2移位到细胞膜。此外,细胞能量消耗的改变,导致细胞损伤,也被发现潜在地破坏细胞的动态平衡。
二、样品处理
关于代谢物的提取,建议根据代谢物的极性指数和logP范围选择提取溶剂。不同极性的混合溶剂在一次提取中可覆盖更多的代谢物,用不同的混合溶剂提取多次可进一步提高提取效率。
近年来发展了许多关于样品制备分析的新方法,如体外液体萃取表面分析(In vitro liquid
extraction surface analysis,ivLESA),可在抽吸培养基后对贴壁细胞直接进行nano-ESI-MS分析。类似的LESA-nano-ESI-MS已被用于细胞脂质组成研究。解吸电喷雾电离(Desorption electrospray
ionization,DESI)能够以最少的制备直接分析生物基质,但局限性之一是非极性化合物的电离效率很低。为了克服这个问题,已经开发了一种基于光电离的正电化组件(DESI/PI)来促进解吸的中性代谢物的二次电离。
最近新型样品基质如胎盘组织、骨骼、视神经、母乳、外淋巴液等已被用于代谢组学,每种基质都需要独特的条件来进行最佳匀质、代谢物提取和分析。
任何仪器技术的进步,如超高效液相色谱或高分辨质谱,都可提供更多和更高质量的信息。
非靶向代谢组学可以作为一种发现和产生假设的工具,其主要目标不是定量,而是以半定量的方式对两个或两个以上的组别进行差异分析。然而,由于无法获得代谢物的定量水平,代谢组学最近的一种趋势是通过靶向方法获得高代谢物覆盖率,这种方法有不同的名字如大规模靶向代谢组学(Large-scale targeted metabolomics),广泛靶向代谢组学(Widely targeted metabolomics)等。
研究者们通常考虑不同方法的组合以获得更高的代谢物覆盖率。当分析物数量增加时,一些化合物可能具有非常相似的结构和相同的质荷比,因此必须进行色谱分离。在这种情况下,通常需要延长色谱分离时间以提高分离度。此外,大多数靶向方法使用三重四极杆,为要测定的不同化合物设置不同的MRM通道,因此代谢物的数量越多,MRM通道数就越多,方法的灵敏度可能会受到影响。有两种方法可以克服这一挑战。一是将一个分析方法中的MRM通道拆分成多个分析方法,一个样品进样多次,但这会影响方法的通量。另一种方法是增加难检测代谢物的MRM通道的驻留时间(Dwell Time),避免创建多个方法。然而,驻留时间的过度增加会使每个峰的点数减少。
除此之外,在一个方法中测定多个代谢物还面临其他两个挑战:定量和克服基质效应。根据是否定量可将代谢组学分为靶向代谢组学和非靶向代谢组学,又可将靶向代谢组学进一步细分为三种策略。一是当有已知浓度或纯度的标准品时,可进行靶向定量分析(Targeted Quantitative Analysis),提供的最终结果是样品中代谢物的真实浓度。使用稳定同位素内标可以克服基质效应,但局限在于可获得的标准品及同位素内标数目有限或价格昂贵。二是当待分析的代谢物标准品无法获得,使用其他结构类似物作为内标的靶向估计分析(Targeted Estimative Analysis),所获得的结果是真正浓度的估计。三是靶向比较分析(Targeted Comparative Analysis),当标准品和结构类似物均无法获得,唯一的选择是比较信号强度,包括处理组样品和对照组样品的比较以及单个样品和混合样品比较。
1.气相色谱-质谱
气相色谱-质谱联用(Gas chromatography-mass spectrometry,GC-MS)是代谢组学中一项非常重要的技术,用于分析易挥发的化合物,但存在峰容量有限,峰重叠严重的问题。在此基础上发展了全二维气相色谱(GC×GC),经第一根色谱柱分离流出的化合物进入第二根色谱柱实现二次分离。基于GC×GC的非靶向代谢组学已用于细菌、尿液、血清和血浆、植物等不同基质,然而GC×GC方法在方法学标准化、数据解析和结果一致性方面仍存在局限,阻碍了该技术充分发挥其潜力。
2.毛细管电泳-质谱
毛细管电泳-质谱(Capillary electrophoresis-mass spectrometry,CE-MS)已被用于代谢组学研究,用于分析各种复杂的生物样品,检测极性和离子化合物。目前,CE−MS被认为是其他技术如HPLC−MS、GC−MS和NMR的补充,进一步提高代谢物的覆盖率。
电喷雾电离(Electrospray ionization,ESI)是最常见的用于CE和MS耦合的软电离技术。然而,CE−MS耦合本身并不简单,它需要毛细管末端的导电液体保持电流并获得兼容的电位,以形成稳定的电喷雾。表1列出了2018年至2019年7月报道的CE−MS在生物样品靶向和非靶向代谢组学中的相关研究。
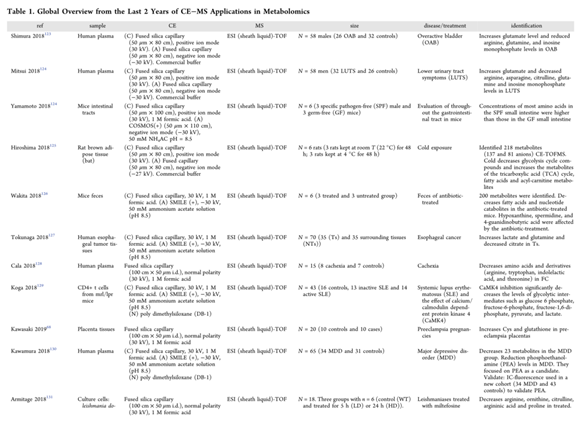
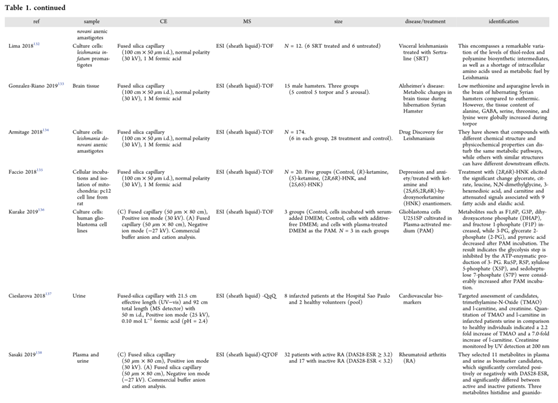

尽管包括大量样本的代谢组学研究的数量有所增加,但这些研究仍然有限。由于CE−MS分离效率很高,非常适合小样本队列的代谢组学研究(≈100),然而,它在大规模人群队列研究中的可靠性仍然存在争议。因此,Harada等人的研究可以被认为是一个里程碑。他们通过分析8413个血浆样本以获得来自日本人群的人类血浆样本代谢图谱,检验了CE−MS在大规模测量中的重复性和有效性。在这项工作中,对80种极性代谢物进行了结构鉴定和定量,在质控样品中的变异系数低于30%。这些结果证明了CE−MS平台在进行大规模队列研究方面的能力。
3.离子迁移谱
在基于质谱的代谢组学研究中,尽管应用不断增加,仪器不断进步,仍有许多因素限制了代谢物鉴定。离子迁移谱(Ion mobility spectrometry,IMS)提供了额外的分离维度,并且工作在毫秒内,可以帮助代谢物鉴定。此外,离子迁移允许测量碰撞截面(Collision cross-section,CCS),这是一个比保留指数更具重现性的正交分析参数,可以和保留时间以及精确分子量信息结合使用,获得代谢物的特性。
近年来,IMS-MS有了相当大的进步,主要分类有漂移时间离子迁移谱(Drift-time ion mobility spectrometry,DTIMS)、行波离子迁移谱(Traveling wave IMS,TWIMS)、高场不对称波形离子迁移谱(High-field asymmetric waveform ion mobility spectrometry,FAIMS或DMS)、捕获离子迁移谱(Trapped IMS,TIMS)等。对于代谢组学分析,TWIMS和DTIMS是最常用的IM类型,FAIMS和TIMS在小分子分析方面也具有巨大的潜力。表2总结了使用不同类型的IMS的最新代谢组学研究。

在所有这些文章中,离子迁移率的加入可以更好地分离共洗脱化合物,降低噪音,更好地分离同分异构体和碎片离子。然而,由于商业仪器相对较新,需要改进软件能力,以便能够通过建立和使用CCS数据库有效地提取多维数据和处理CCS值。
4.手性分析
许多生化过程不仅包括内源化合物,还包括外源化合物和微生物代谢物等,表现出立体特异性,这些类型化合物的分析需要有效的分离方法。由于样品的复杂性和对映体的分离,二维液相色谱(Two-dimensional liquid chromatography,2D-LC)特别适合手性代谢组学,但主要缺点是数据管理和分析时间长(>1h)。
关于手性代谢组学的靶向或非靶向的方法,通常基于一组具有相同官能团的化合物或者和某条代谢途径相关的代谢物。非靶向或大规模的手性代谢组学研究在文献中并不常见。手性代谢物的准确定量需要使用内标来克服基质效应引起的离子抑制,可使用同位素标记衍生化的方法,目前已开发了很多手性或非手性同位素标记衍生化试剂。
四、数据处理
由质谱产生的数据是庞大且复杂的,因此需要大量的算法和工作流程处理和分析获得的大量信息。图1展示了代谢组学数据特征及数据处理策略。
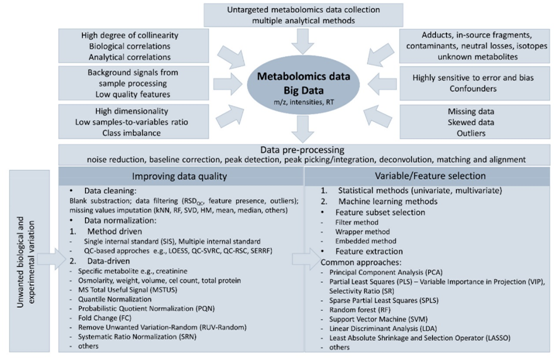
图1.代谢组数据组成、数据质量提高策略和变量选择步骤
1.提高数据质量
数据清洗
高质量数据的重要性不言而喻,因此,代谢组学数据清洗是必要的。这种策略的主要好处是为相关特征建立数据归一化参数。综合的数据预处理和降噪算法以及多种数据筛选策略可以有效地提高总体数据质量。
数据标准化
虽然数据标准化的概念是已知的,但在代谢组学数据背景下,仍不够明确。考虑到可用算法的多样性以及关于系统偏差来源的有限信息,为给定的数据集选择最佳方法是一项重大挑战,也是目前代谢组学数据预处理的主要障碍。已经提出了几种基于不同原理的标准化策略,这些策略旨在消除由实验源(例如,实验偏差、信号漂移、样品降解、测量变化、进样体积变化和样品提取效率)和生物变异(例如,生物体液浓度变化、不同细胞大小和不同样品测量)引起的混淆。经常在基于MS的非靶向代谢组学中观察到仪器响应的系统性变化,批次内和批次间效应都会降低统计效能,并对报告结果的重复性和再现性产生负面影响,阻碍数据解释。然而,LC-MS代谢组学数据暴露于由以下原因引起的特定的变异源:(I)MS源中污染物的累积,(II)源自流动相组成和制备的杂质或变异性,(III)分析柱性能,或(IV)样品基质复杂性的差异,因此,需要更有效的批次校正策略。在假设QC响应与进样顺序无关的情况下,基于QC重复分析的算法是最推荐的方法之一。
任何校正方法都应谨慎使用,因为可能会严重影响潜在生物标志物的选择,并可能导致错误的结论。因此,最优方案的选择必须依赖于数据,满足实验设计的标准和研究目的。
2.变量/特征选择
生物标志物的主要作用是区分不同的组别,例如,健康和疾病。因此,选择携带相关生物信息的变量是代谢组学工作流程中最重要和最具挑战性的任务之一。目前已经开发了许多单变量或机器学习方法,如聚类、回归或分类,以帮助将组学数据转化为实际的临床应用。这些方法可以分为(I)特征子集选择方法(删除不起作用或多余的变量)和(II)特征提取方法(创建原始变量的组合作为新特征,以降低所选变量的维度)。表3提供了相关算法的概述。此外,特征选择可以使用非监督模型或监督模型。多变量分析方法,如无监督主成分分析或有监督偏最小二乘法,已成为代谢组学的标准程序。然而,最近在代谢组学研究中报道了越来越多的嵌入式方法,如随机森林(RF)、神经网络或最小绝对值收敛和选择算子(LASSO)方法,以及许多其他基于机器学习算法的方法。

五、代谢物注释和鉴定
根据所采用的分析技术,代谢物鉴定工作流程存在较大差异。
1.GC-EI-MS
GC-MS通常配备EI作为电离源,分子碎裂模式高度可重复。此外,根据保留时间可以很容易地计算保留指数。目前基于GC-MS数据的代谢物鉴定有较完善的方法,GC-MS数据库包含单同位素质量、碎裂模式和保留指数等信息。
代谢物注释通常使用商业化数据库,如NIST,Wiley,MoNA和Fiehn;开源数据库,如GOLM或者自建库。基于GC-MS的非靶向代谢组学在代谢物注释方面已经取得了重要进展,但在未知代谢物鉴定方面仍有许多目标需要实现。
2.LC-ESI-MS和CE-ESI-MS
在代谢物鉴定方面,CE-MS和LC-MS的重现性不如GC-MS,这主要是由于ESI源可能产生加合物、二聚体和碎片(包括源内裂解),导致了一种本质上不同的代谢物鉴定方法。近年来,代谢组学许多化学信息学和生物信息学工具已经取得了巨大进步,为阐明未知化合物结构提供了有力支持(如图2所示)。
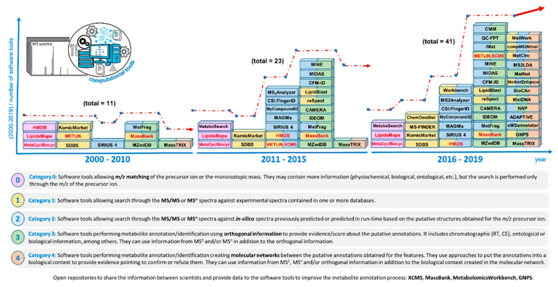
图2.代谢物注释和鉴定的时间轴及软件工具的开发
专门用于代谢组学研究的数据库在21世纪初创建,最初只提供化合物的m/z和结构(The Cyc Collection,Metlin,LIPID MAPS,HMDB,或KomicMarket)(见图2,第0类)。然而,实验测到的化合物数量并不能覆盖完整的代谢组,因此,开发了一些从已知的代谢物生成合理结构的工具(MINE和BioTransformer)。
代谢组数据库开发的下一阶段是包括碎片相关的信息,允许搜索谱图(XCMS,HMDB或MassBank)(见图2,第1类)。然而,由于标准品的数量有限,不可能获得这些数据库中存在的大多数化合物的实验数据。因此,创建了不同的工具和方法来预测不同实验条件下的碎裂谱图(MetFrag,MAGMa,MyCompoundID,CFM-ID,或CSI:FingerID)(见图2,第2类)。
由于相似的结构通常具有相似的碎裂模式,代谢物的鉴定仍然缺乏信心,因此使用正交信息注释和鉴定代谢物。例如,化合物的疏水性决定了它在色谱柱上的保留时间或毛细管电泳中的迁移时间。应用正交信息注释代谢物的另一个例子是评估一种化合物可能形成的离子(加合物、多电荷、二聚体等)。这种正交信息已逐渐包含在代谢物注释和识别工具(MZeDB,CAMERA,MetFrag,LipidBlast,iMeT,CMM)中(见图2,第3类)。
最近,开发了一些包括生物体不同代谢物之间生物关系的工具(MassTRIX,GNPS,xMSannotator,BioCAn,NAP,ADAPTIVE,MetDNA、MolNetEnhancer,或MetNet)(见图2,第4类)。
代谢物注释和鉴定数据库逐渐包含了更多关于化合物的信息。研究代谢物网络和评估推定注释之间的关系是最近创建或更新工具的发展趋势。当使用更多的信息进行代谢物注释时,代谢组学实验将获得更高的置信度。
此外,可用于进行代谢物注释和鉴定的大量工具(本综述包括41个,但这个数字正在指数级增加)使得用户需要学习多种不同语言,如R、Python、Web应用程序和独立应用程序,因此出现了集成代谢组学实验所有阶段使用的不同工具的框架或工作流(Workflow4Metabolomics,Taverna和KNIME)。
同时,开源数据库(XCMS、MassBank、MetabolomicsWorkbench和GNPS)的存在使研究人员能够传播获得的信息,与代谢组学领域的其他研究者共享。这些数据库承诺以更标准化的方式呈现结果,并有机会在后续实验中使用其他研究人员的数据来改进注释和鉴定。然而,大量用户上传关于相同代谢物的相同或非常相似的信息可能会使数据难以使用。由于专门用于不同代谢物类型、方法或目的的数据库之间重叠较少,用户友好和免费访问不同的数据源至关重要。
结论
分析化学的概念正在发展,包括非靶向、差异分析和半定量分析。在进行差异分析时,会通过统计分析来消除可能影响所有比较样本的因素,从这个意义上说,它们的严格控制并不关键。同时,这并不意味着代谢组学可以被用作一个黑匣子,在黑匣子里输入样本,按下仪器上的按钮,软件平台自动提供结果。在处理未知因素时,深入了解分析的目标、样本、分析工具,以及数据挖掘和代谢物鉴定策略,对于获得有意义的结果至关重要。







